
Design changes in medical devices could be modifications made to the design, materials, functionality, or intended use of the device. Moreover, changes in medical device design, manufacturing processes, or regulatory requirements are inevitable in the industry. Hence, change control is vital to ensure the changes do not compromise safety, performance, or regulatory compliance of the device. Without proper medical device design change management, companies risk safety concerns, regulatory violations, and product recalls. Hence, effective change control is vital for maintaining compliance with ISO 13485 design changes and FDA 21 CFR Part 820.30(i).
What is ISO 13485 design changes?
ISO 13485 design changes are modifications made to the design and development of medical devices. These changes can cover changes in materials, software, functionality, or intended use. All of these are done while ensuring compliance with regulatory requirements. Design changes in medical devices can be done for various reasons. These changes can be aimed at performance improvements, regulatory compliance, and usability enhancements.
According to ISO 13485 design changes requirements, manufacturers are required to establish a robust system to control design changes. Hence, establishing a change control system that defines responsibilities, timelines, and approval processes for modification is vital.
Examples of design changes in medical devices
- User interface modification: Changes can be done to the device’s display, controls, or software interface to improve its usability and patient interaction.
- PCB layout changes: Manufacturers may modify the printed circuit board (PCB) layout to improve device efficiency or accommodate new components.
- Adding new features or refining existing ones: Manufacturers can improve the functionality of the medical device by adding new features or refining existing ones.
- Material changes: Manufacturers may change the materials used in the device to improve cost-effectiveness, durability, and biocompatibility.
Regulatory considerations for medical device design change management
In addition to ISO 13485 design changes guidelines, medical device manufacturers will have to adhere to FDA 21 CFR Part 820.30(i). This regulatory guideline requires medical device manufacturers to establish procedures for identifying, documenting, validating, and approving design changes before implementing them.
Need for change control for medical devices
The medical device industry is strictly regulated for quality, safety, and efficacy. Hence, change control is of paramount importance in medical device industry. This is because any design change not managed properly will affect product performance, thereby adversely impact patient safety and outcome. We have highlighted the need for a proper change control system.
- Regulatory compliance: A robust medical device change control system ensures adherence to ISO 13485, FDA 21 CFR Part 820, and other global regulatory standards.
- Risk management: Unintended consequences of device design changes that could lead to device failures can be avoided. It helps with risk assessment and validation before implementation of changes.
- Traceability: It helps maintain detailed records of design changes. This ensures traceability and audit readiness for regulatory inspections.
- Validation of changes: The medical device change control system will ensure the changes do not introduce new risks or adversely affect device functionality.
- Proper documentation: Proper documentation of all changes in medical devices.
5 key steps in managing ISO 13485 design changes
1. Initiating a medical device design change request
- Description of the proposed changes.
- Justification and reason for the change.
- Assessment of the impact of the changes on performance, safety, and regulatory compliance of the device.
2. Medical device design change review and approval process
- Design engineering assessing the feasibility and technical impact of the change
- Evaluation of operational impact of the change, specifically on production and supply chain.
- Quality assurance system for reviewing risk and validation requirements.
- Compliance with applicable regulatory standards has to be ensured at all stages.
3. Risk and regulatory assessment of the changes
- Assessment of product safety and usability after implementation of proposed changes
- Evaluation of labelling updates that do not require major design alterations
- Checking for regulatory updates requiring modifications of the device
4. Verification and validation of changes
- Verification of the modifications ensures the change meets design specifications
- Validation confirms the change does not adversely impact performance or safety of the device
- Testing and clinical evaluations may be required for high-risk modifications done on medical devices.
5. Proper documentation has to be maintained to ensure traceability of the ISO 13485 design changes
- Design change request (DCR) form has to be duly filled
- Risk assessment reports have to be prepared
- Verification and validation of the changes have to be properly recorded
- Design files and device master file (DMF) have to be updated
- If required, regulatory submissions have to be prepared for compliance
Best practices for managing ISO 13485 design changes
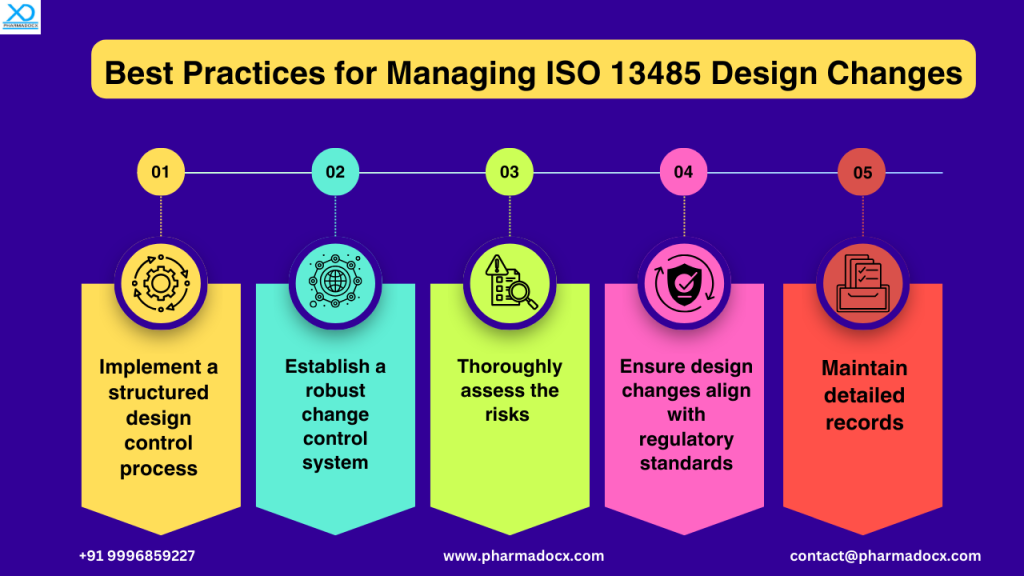
Regulatory requirements for ISO 13485 design changes
ISO 13485 design changes guidelines require manufacturers to establish a robust system to control design changes. The requirements are:
- Identification and documentation of all design changes.
- Verification and validation before implementation of changes.
- Formal approval and proper record keeping for traceability of changes.
- Review of potential impact on safety, usability, and regulatory compliance of the device.
- Perform evaluations to ensure modifications do not introduce new risks.
- Ensure changes are reviewed and authorized before implementation.
FDA 21 CFR Part 820.30(i) guidelines for medical device design changes
FDA has formulated certain regulations for medical device design change management. Failure to comply with these guidelines may result in FDA warning letters, recalls, or regulatory sanctions and fines. As per FDA regulations, medical device manufacturers are required to comply with the following guidelines:
- Identify, document, and assess changes in medical device designs
- Conduct risk-based validation or verification of changes
- Maintain records of all ISO 13485 design changes and modifications.
- Ensure changes are reviewed and approved prior to their implementation.
How can Pharmadocx Consultants help you comply with ISO 13485 design changes guidelines?
We at Pharmadocx Consultants have extensive regulatory expertise and industry experience. Our team will help you manage design changes while ensuring compliance with ISO 13485 and FDA 21 CFR Part 820.30 in a hassle-free manner. Our service scope covers the following:
- Change control implementation: We will help you establish a robust and well-structured system for reviewing, approving, and documenting design modifications of the medical device.
- Validation and verification: We will help you thoroughly assess changes to evaluate their impact.
- Proper documentation: We provide documentation support. We will help you record design updates and modifications.
- Risk assessment: We will help you perform risk assessments to identify potential risks and formulate mitigation strategies.
- Regulatory compliance: Ensuring modifications in the medical device comply with ISO 13485 design changes and FDA 21 CFR Part 820.30 guidelines.
Drop an email at [email protected] or call/Whatsapp on 9996859227 and our team of experts will get back to you.


